Customizable Synthetic Antibiotic Outmaneuvers Resistant Bacteria
Researchers’ Molecular ‘LEGO Set’ Advances Efforts to Defeat Resistant Microbes
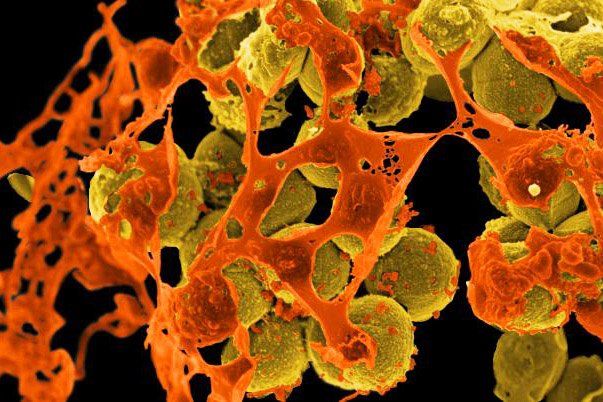
Scanning electron micrograph of methicillin-resistant Staphylococcus aureus (MRSA, brown) surrounded by cellular debris. MRSA resists treatment with many antibiotics. Image by NIAID
Antibiotic resistance is one of the world’s most urgent public health threats. In the United States alone, tens of thousands of deaths result each year from drug-resistant strains of common bacteria such as Staphylococcus aureus and Enterococcus faecium, which can cause virtually untreatable hospital-acquired infections. Perilously few new classes of antibiotics are being developed to fight infections that have become resistant to traditional treatments, and bringing any new drugs to market could take decades.
Researchers at UC San Francisco are tackling antibiotic resistance using a different approach: redesigning existing antibiotic molecules to evade a bacterium’s resistance mechanisms. By devising a set of molecular LEGO pieces that can be altered and joined together to form larger molecules, the researchers have created what they hope is the first of many “rebuilds” of drugs that had been shelved due to antibiotic resistance. The research was published Sept. 23 in Nature.
“The aim is to revive classes of drugs that haven’t been able to achieve their full potential, especially those already shown to be safe in humans,” said Ian Seiple, PhD, an assistant professor in the UCSF School of Pharmacy’s Department of Pharmaceutical Chemistry and the Cardiovascular Research Institute (CVRI), and lead author on the paper. “If we can do that, it eliminates the need to continually come up with new classes of drugs that can outdo resistant bacteria. Redesigning existing drugs could be a vital tool in this effort.”
In work descibed in the new Nature paper, Seiple and his collaborator James Fraser, PhD, a professor in the School of Pharmacy’s Department of Bioengineering and Therapeutic Sciences, have demonstrated this approach with a class of antibiotics called streptogramins. Until recently, streptogramins were very effective against S. aureus infections, until the bacteria evolved a clever resistance mechanism.
Streptogramins disable bacteria by gumming up the works in the bacterial ribosome, making it impossible for the bacteria to make proteins. But bacteria resistant to streptogramins produce proteins called virginiamycin acetyltransferases (Vats), which recognize these antibiotics when they enter the bacterial cell. The Vats grab the drug and chemically deactivate it before it can bind to the ribosome, rendering it useless.
Streptogramins, like most other antibiotics, are derived from naturally occurring antibiotic compounds produced by other organisms (usually bacteria) that are then tweaked to optimize their performance in the human body. Seiple figured that there must also be a way to make further changes to the drug molecule that would allow it to evade capture by the Vat proteins.
Seiple set out to build new streptogramins from the ground up, rather than modifying existing structures. To make the building process easier, Qi Li, PhD, a postdoctoral fellow in the Seiple lab and co-first author on the paper, created seven molecular modules that can be tweaked as needed to build a set of variations on the streptogramin molecule.
“This system allows us to manipulate the building blocks in ways that wouldn’t be possible in nature,” said Seiple. “It gives us an efficient route to re-engineering these molecules from scratch, and we have a lot more latitude to be creative with how we modify the structures.”
Once Seiple and Li had their building blocks, the next step was to get a molecular-level view of the chemistry involved in order to better understand how to modify and piece together those molecular LEGOs.
For that, Seiple teamed up with Fraser, who specializes in creating visual models of biological molecules.
“My lab’s contribution was to say, ‘Now that you've got the seven pieces, which one of them should we modify and in what way?’” said Fraser, whose work on the project was supported by the inaugural Sanghvi-Agarwal Innovation Award.
To get answers to that question, Jenna Pellegrino, a graduate student in the Fraser Group and co-first author on the paper, used two complementary techniques, cryo-electron microscopy and x-ray crystallography, to create three-dimensional pictures of the drug at near-atomic resolution, as well as its target the bacterial ribosome, and its nemesis, the Vat protein.
Using the models, Li, Pellegrino, Seiple and Fraser could see which parts of the streptogramin molecule are essential to the antibiotic’s function. Then Li was free to fiddle with the drug’s non-essential regions to find modifications that prevented Vats from interacting with the drug while still allowing it to bind to its ribosomal targets and disable the bacterium.
The team found that two of the seven building blocks seemed to offer potentially interesting sites for modification. They made variations of the drug that contained tweaks in those regions and found that these variations had activity in dozens of strains of pathogenic bacteria. The researchers also tested their most promising candidate against streptogramin-resistant S. aureus in infected mice, and found it was over 10 times more effective than other streptogramin antibiotics.
Seiple points out that the knowledge gained through these collaborative experiments can be applied to modifying many other antibiotics.
“We learned about mechanisms that other classes of antibiotics use to bind to the same target,” he said. “In addition, we established a workflow for using chemistry to overcome resistance to antibiotics that haven't reached their potential.”
Seiple will continue to refine these synthetic streptogramins and then hopes to move the work to the private sector where the reengineered antibiotics could be further developed and tested in human trials. He and Fraser plan to continue working together on reviving other antibiotics that have been shelved because of microbial resistance, refining a set of tools that can help researchers stay one step ahead of bacterial evolution.
“It’s a never-ending arms race with bacteria,” said Fraser. “But by studying the structures involved—before resistance arises—we can get an idea of what the potential resistance mechanisms will be. That insight will be a guide to making antibiotics that bacteria can’t resist.”
Authors: The study was led by Ian Seiple of UCSF. Qi Li, Jenna Pellegrino, D. John Lee, Arthur Tran, Hector A. Chaires, Ruoxi Wang, Jesslyn E. Park, Kaijie Ji, David Chow, Justin T. Biel, Matthew Jacobson, and James Fraser of UCSF; Axel Brilot and Kenneth Borrelli of UCSF and Howard Hughes Medical Institute; Na Zhang of UCSF and Beijing University of Technology; Gydo van Zundert of Schrödinger, Inc.; Dean Shinabarger, Cindy Wolfe, and Beverly Murray of Micromyx; and Estelle Mühle and Olivier Chesneau of Institut Pasteur are also co-authors.
Funding: This project was funded by the UCSF Program for Breakthrough Biomedical Research, funded in part by the Sandler Foundation (J.S.F. and I.B.S.), a Sanghvi-Agarwal Innovation Award (J.S.F.), Packard Fellowships from the David and Lucile Packard Foundation (J.S.F. and I.B.S.), NIH GM123159 (J.S.F.), and NIH GM128656 (I.B.S.)
Disclosures: Borrelli and van Zundert are employees of Schrodinger Inc. Shinabarger, Wolfe and Murray are employees of Micromyx.
The University of California, San Francisco (UCSF) is exclusively focused on the health sciences and is dedicated to promoting health worldwide through advanced biomedical research, graduate-level education in the life sciences and health professions, and excellence in patient care. UCSF Health, which serves as UCSF’s primary academic medical center, includes top-ranked specialty hospitals and other clinical programs, and has affiliations throughout the Bay Area.