UCSF Researcher's Drug Strategy for Huntington's Disease
By Jeffrey Norris
Twice a month, UCSF neurologist Marc Diamond, MD, runs one of the only Huntington's disease (HD) clinics in Northern California.
Huntington's is insidious. Most often, it is not diagnosed until midlife. The emergence of symptoms - including impaired movement, thought and mood - is a harbinger of disease progression that leads eventually to premature death. Diamond and his clinical colleagues treat HD symptoms, but physicians have nothing to offer yet to halt the onset or progression of the disease.
The devastation caused by this degenerative brain disorder is a constant motivator in Diamond's research. He aims to get into clinical trials new treatments that may stop the molecular and cellular events that drive Huntington's.
Based on his discoveries from studies of molecules, cells, flies and mice, Diamond believes he is on track to spark development of one of the first effective treatments for the disease. He invented an efficient way to screen for candidate drugs that block what may be the biochemical trigger for Huntington's.
Diamond identified a drug prototype by using the novel screening method. The prototype appears to relieve symptoms in animal models of Huntington's disease. Diamond's work may pave the way for the development of newly formulated pharmaceuticals based on fundamental disease mechanisms.
Huntington's Is a Genetic Disease
Despite its delayed onset, Huntington's is a genetic disorder passed from parent to offspring. Inheriting just one mutated copy of a specific gene from an affected parent guarantees that HD will strike. Each child of a person who carries the HD gene will have a 50-50 chance of also inheriting the gene and suffering the disease. Parents may pass on the HD gene even before symptoms reveal that they themselves carry it. An estimated one in 10,000 individuals in the United States carries the gene, while approximately 30,000 people are symptomatic.
A genetic test for the HD gene has been available since 1993. Those who test negative know they will remain free from the disease. Those who test positive may be able to better plan for the future. But 14 years after the mutated gene that causes HD was identified, the role of the protein it encodes and its connection to disease progression remain unclear and debated among researchers. The discovery of the gene has not yet led to any therapeutic benefit.
Huntington's Protein Molecules Clump Together
Based on evidence from his own lab and from the labs of others, Diamond believes the key to stopping HD is to stop HD proteins from undergoing a pathological shape change and sticking together. The abnormal HD gene causes the encoded protein to misfold and assume a shape that results in the protein molecules aggregating with others of their own kind, Diamond says.
Like all proteins, the protein that is abnormal in HD is made from 20 different amino acid building blocks. The amino acids in proteins are linked in chains. The chains fold into three-dimensional shapes that are distinct for different proteins.
The HD protein is one of many known to contain 10 or more consecutive molecules of the amino acid glutamine linked end to end within the protein. The function of the glutamine "repeats" in these proteins is not clear. The number of glutamine repeats in the HD protein varies from person to person. When the number of glutamine repeats in the Huntington's protein exceeds 36, the disease may strike.
Mysteriously, individuals with 35 or fewer glutamine repeats in the HD protein never get Huntington's. Those with 40 or more glutamine repeats always get the disease. Diamond was fascinated by this phenomenon when he learned of it more than a decade ago, and decided to conduct research in addition to practicing neurology.
There are now nine known diseases caused by proteins with expanded glutamine repeats, Diamond says. Each affects a distinct population of nerve cells and causes a different degenerative disease in the brain.
Medical researchers debate the role of the HD protein in causing Huntington's. Some say an inability of the abnormal protein to perform its normal function is to blame. Others suggest that single molecules of the abnormal protein are somehow toxic.
Diamond believes instead that the disease is driven by the aggregation of abnormal copies of the protein within certain nerve cells, which somehow disrupts and eventually kills them. This is likely to be a common mechanism behind all the diseases associated with expanded glutamine repeats, according to Diamond. What causes the aggregation is misfolding of the protein triggered by too many glutamine repeats, he suggests.
"I see nine different genes for proteins with completely different functions and structures," Diamond says. "All they have in common is that they all have glutamine repeats, they all cause a degenerative syndromes, and the syndromes all share common pathological features, including protein aggregation. The numbers of glutamine repeats that result in disease also are similar. The simplest interpretation is that the repeats produce a common, abnormal toxic structure."
In most common degenerative brain diseases that do not involve glutamine repeat proteins - such as Alzheimer's and Parkinson's - other proteins also clump together in nerve cells, Diamond notes.
"The brain seems to be especially vulnerable to proteins that tend to misfold," he says. "Virtually all the major neurodegenerative diseases - and rare ones too - are associated with misfolded proteins that accumulate in cells within the brain."
Shedding Fluorescent Light on Huntington's
Diamond invented a new protein-tracking method based on a technique called fluorescence resonance energy transfer (FRET). With the new method, he can quickly measure how much HD protein is aggregating in cells that he grows in the lab.
Diamond labels the HD protein with fluorescent tags. He fine-tunes a specialized fluorescent-light "reader" to detect a distinctive light pattern when the HD protein aggregates. Diamond can use the technique to quantify protein aggregation even when the aggregates are too small to view in a pathologist's microscope.
Using FRET, Diamond efficiently screened thousands of drugs and other molecules of known biological activity. He has determined which ones best prevented aggregation of the HD protein within cells. The researchers more extensively studied one molecule called Y-27632. This chemical reduced HD protein aggregation by 20 percent. Y-27632 targets a regulatory protein that controls the cell's internal scaffolding, called the actin cytoskeleton.
Aiming for Huntington's Treatment
When Diamond treated flies with neurodegeneration caused by the glutamine repeats, the flies lost fewer nerve cells in their eyes. More recently, Diamond has been working with mice genetically engineered to make the human HD protein. Mice with the resulting disease have difficulty keeping their balance. Diamond has treated these mice with Y-27632 and has reported preliminary, promising results at recent scientific meetings.
The HD protein does not simply have a propensity to associate with others of its kind, Diamond says. His lab studies indicate that other molecules - such as the protein targeted by Y-27632 - are involved in the biochemical regulation of HD protein aggregation. The quantitative FRET assay permits Diamond to identify other molecules that may help bring HD proteins together or keep them apart. Some of these molecules might also eventually prove to be promising drug targets to block the mechanisms of HD and other glutamine-repeat diseases.
"I am hopeful that we can start testing the first mechanism-based therapies for HD within several years," Diamond says.
Related Links:
Diamond Lab
A Rapid Cellular FRET Assay of Polyglutamine Aggregation Identifies a Novel Inhibitor
Neuron, November 13, 2003
Institute for Neurodegenerative Diseases
UCSF Memory and Aging Center
Huntington's Disease
National Institute of Neurological Disorders and Stroke, NIH
Huntington's Disease Society of America
Hereditary Disease Foundation
 |
Marc Diamond |
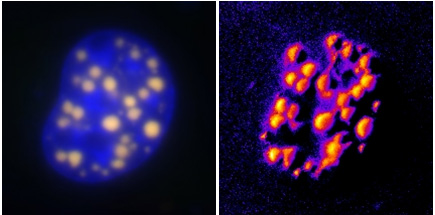 |
On the left is a conventional fluorescent microscopy image highlighting aggregations of the protein that is abnormal in Huntington's disease (HD). On the right, the new FRET method developed by Marc Diamond of UCSF highlights the HD protein with greater sensitivity. |
Related Links
- Diamond Lab
- A Rapid Cellular FRET Assay of Polyglutamine Aggregation Identifies a Novel Inhibitor
- Neuron, November 13, 2003
- Institute for Neurodegenerative Diseases
- UCSF Memory and Aging Center
- Huntington’s Disease
- National Institute of Neurological Disorders and Stroke, NIH
- Huntington’s Disease Society of America
- Hereditary Disease Foundation